Primer (PCR)
Introduction
The idea of the polymerase chain reaction, PCR (Polymerase Chain Reaction) was developed in the 1980s by Kary Mullis. With this technique, he revolutionized molecular biology and for this reason won the Nobel Prize.
PCR is an in vitro technique, used to enzymatically amplify a specific region of deoxyribonucleic acid (DNA). The DNA region of interest is bounded by two initiators (“primers”), which are short fragments of DNA. This reaction can start from a single strand of DNA, which is amplified until millions of copies of the original are obtained in just a few hours.
To improve the specificity of the reaction, the initiators must be carefully selected and in this way reduce the risk of dimer formation and non-specific products that would alter the result.
PCR
PCR repeatedly uses high temperatures to denature the DNA template or separate its strands.
The DNA polymerase normally used in PCR is called Taq polymerase, after the heat-tolerant bacterium from which it was isolated (Thermus aquaticus).
T. aquaticus lives in hot springs and hydrothermal vents. Its DNA polymerase is very thermostable and its greatest activity occurs near 70º C (a temperature at which the DNA polymerase of humans or E. coli would not work). Taq polymerase is ideal for PCR thanks to this thermal stability.
Primer
Like other DNA polymerases, Taq polymerase can only make DNA if there is a primer, a short sequence of nucleotides that provides a starting point for DNA synthesis. In a PCR reaction, the region of DNA that will be copied, or amplified, is determined by the primers that are chosen.
PCR primers are short pieces of single-stranded DNA, generally about 20 nucleotides in length. In each PCR reaction, two primers are used that are designed to flank the target region (the region that must be copied). That is, sequences are added to them that will make them bind to opposite strands of the DNA template only at the ends of the region to be copied. The primers bind to the template through base complementarity.
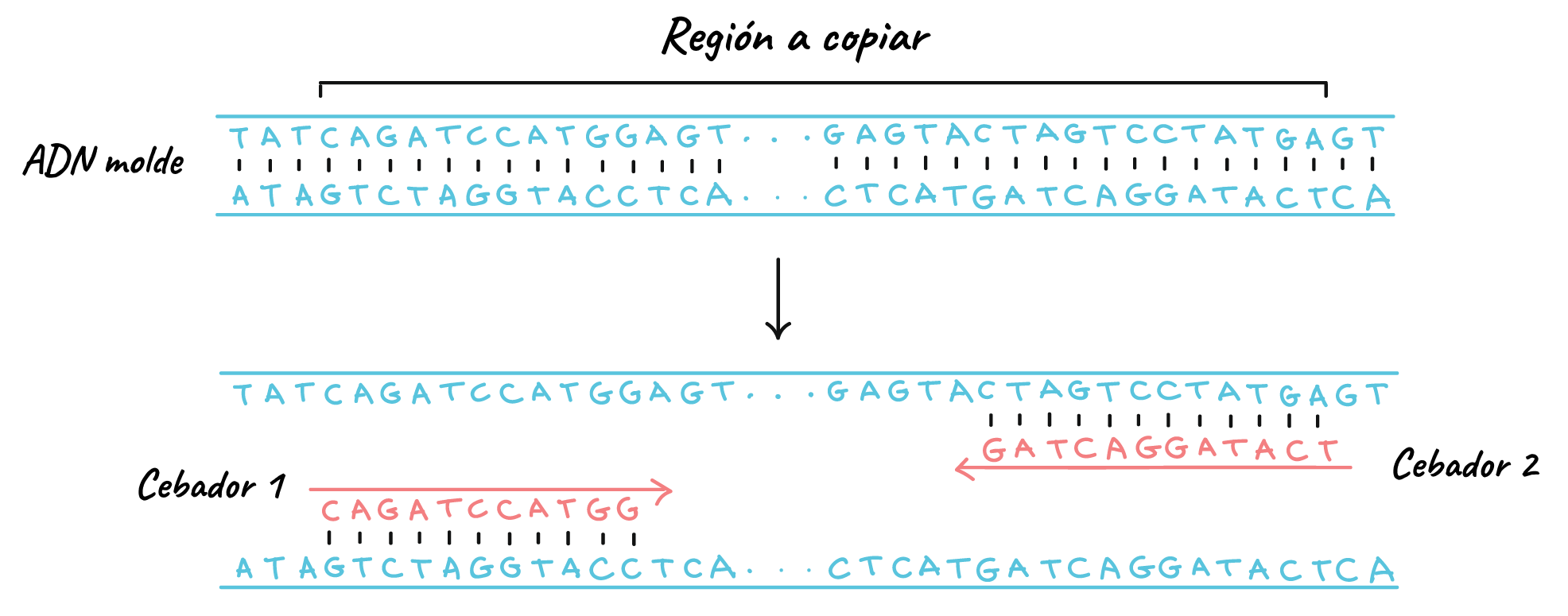
When the primers bind to the template, the polymerase extends them and the region between them is copied.
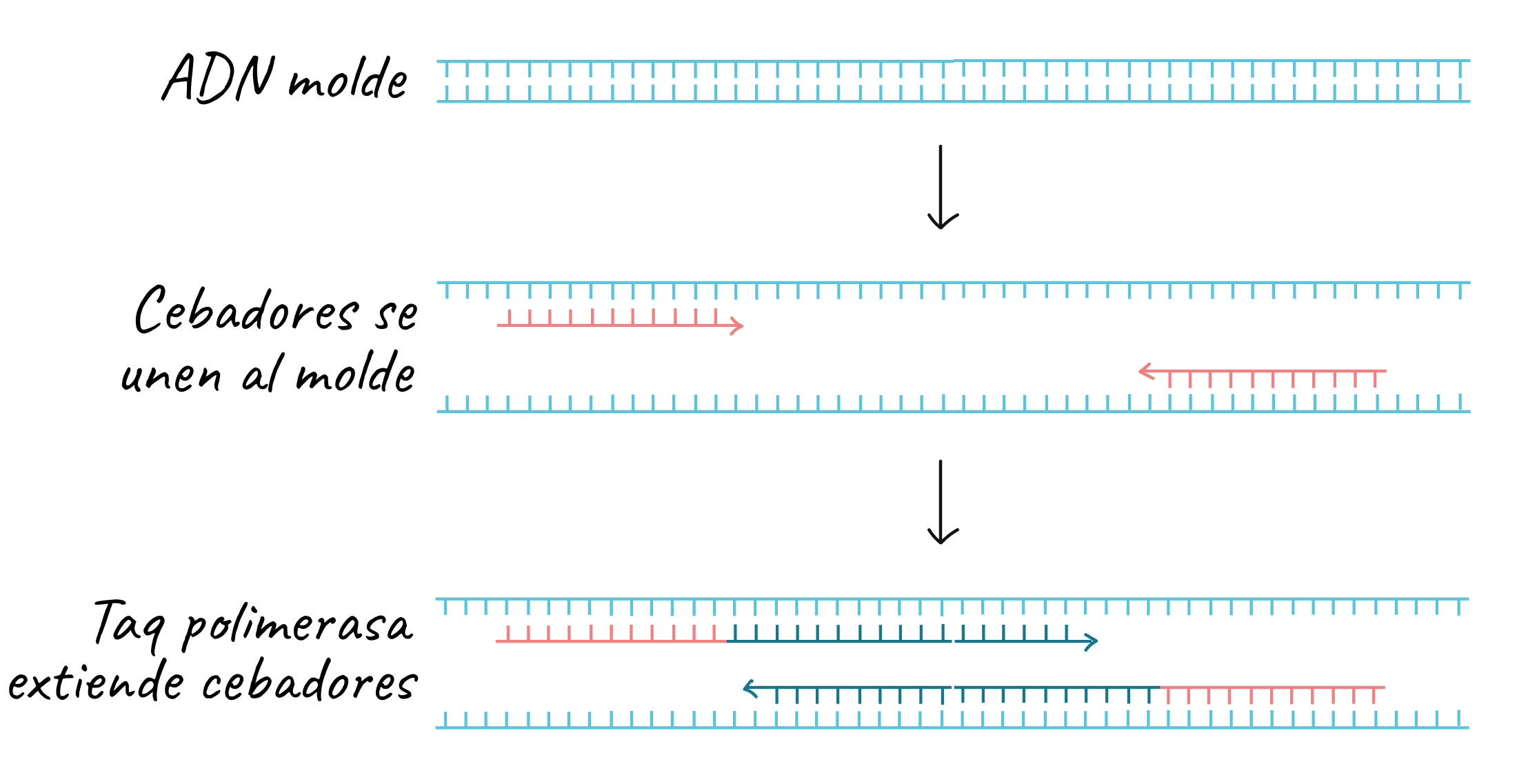
Both primers point “inward” when they bind, that is, in the 5’ to 3’ direction toward the region to be copied. Like other DNA polymerases, Taq polymerase can only synthesize DNA in the 5’ to 3’ direction. As the primers extend, the region between them is copied.

Steps
The key ingredients for a PCR reaction are Taq polymerase, primers, template DNA and nucleotides (the building blocks of DNA). The ingredients are placed in a tube, together with the cofactors that the enzyme needs, and are subjected to repeated cycles of heating and cooling that allow DNA synthesis.
The basic steps are:
-
Denaturation (96 ºC): the reaction is heated considerably to separate, or denature, the DNA strands. This provides the single-stranded templates for the next step.
-
Annealing (55 - 65 ºC): the reaction is cooled so that the primers can bind to their complementary sequences on the single-stranded DNA template.
-
Extension (72 ºC): the temperature of the reaction is raised so that the Taq polymerase extends the primers and thus synthesizes new DNA strands.
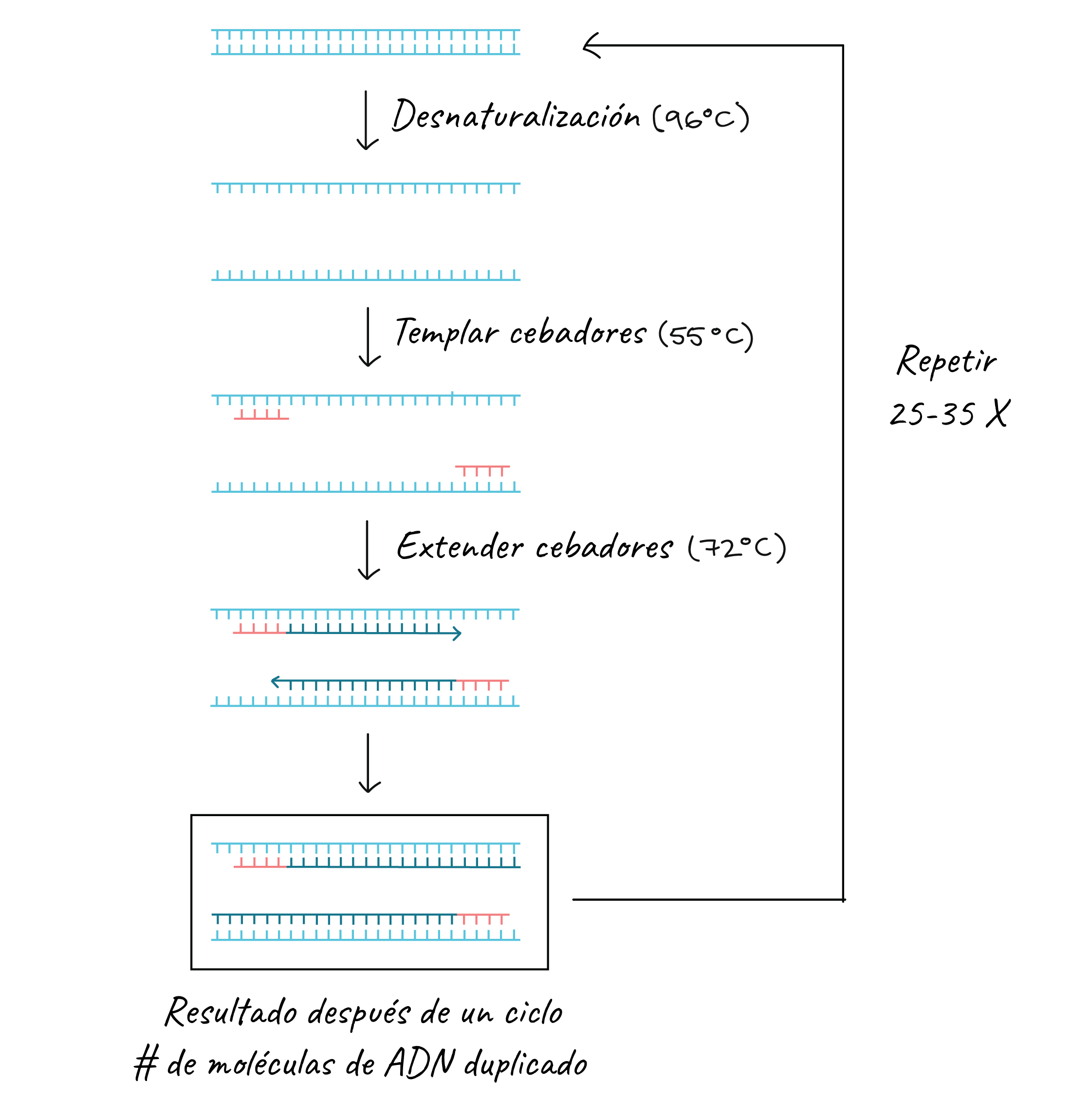
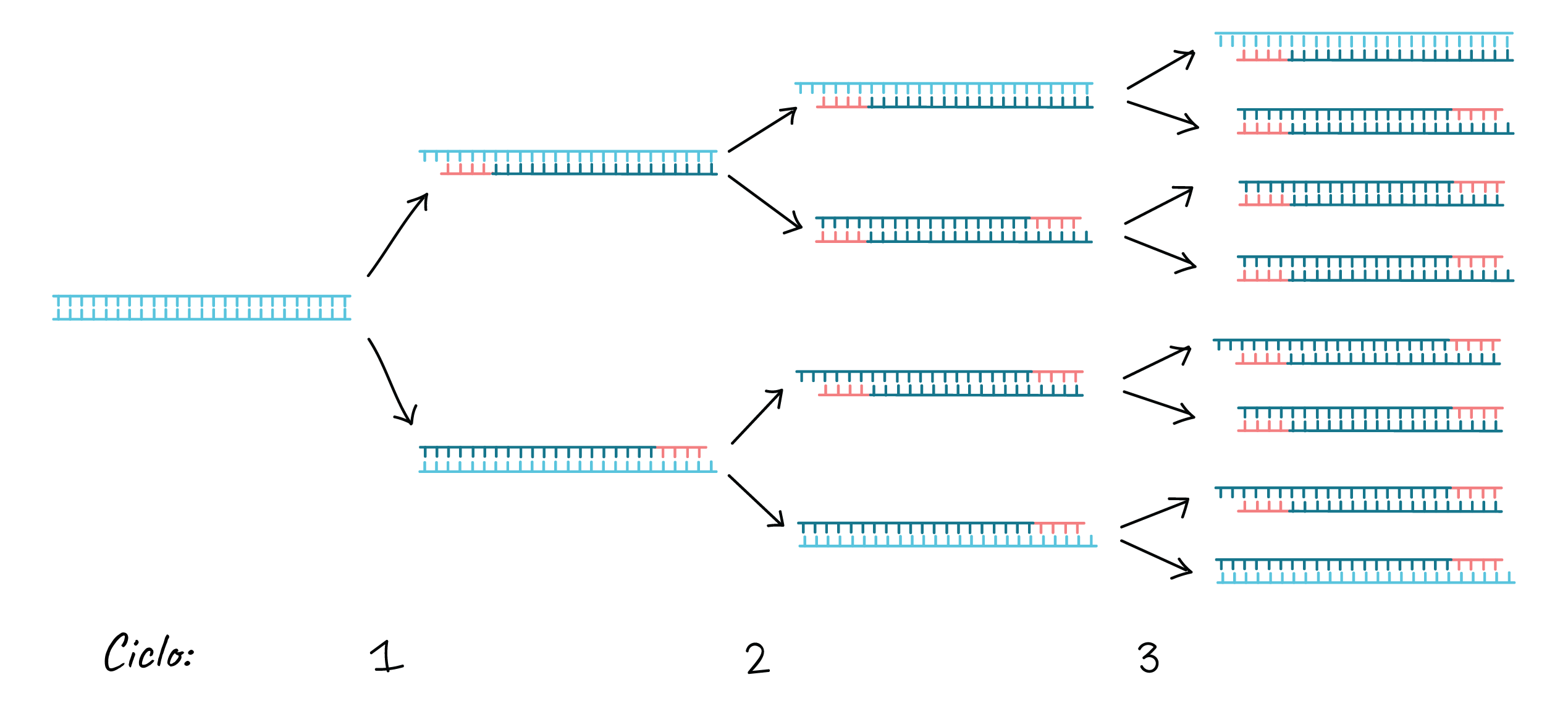
This cycle is repeated 25 - 35 times in a typical PCR reaction, which generally takes 2 - 4 hours, depending on the length of the DNA region being copied. If the reaction is efficient (works well), it can produce billions of copies from one or a few copies of the target region.
That is because not only is the original DNA used as a template in each cycle. In reality, the new DNA produced in one round can serve as a template in the next round of DNA synthesis. There are many copies of the primers and many Taq polymerase molecules floating in the reaction, so the number of DNA molecules can almost double in each cycle. The following image shows this exponential growth pattern.
Primer design
The main property of primers is that they must correspond to sequences of the template molecule (they must be complementary to the template strand). However, it is not necessary for the primers to fully correspond to the template strand; the only essential thing is that the 3’ end of the primer corresponds completely to the template DNA strand so that elongation can continue.
A guanine or cytosine is generally used at the 3’ end, and the 5’ end of the primer usually has stretches of several nucleotides. In addition, both 3’ ends of the hybridized primers must point toward each other.
The size of the primer is also very important. Short primers are mainly used to amplify a small and simple DNA fragment. On the other hand, a long primer is used to amplify a sample of eukaryotic genomic DNA. However, a primer should not be too long (primers > 30 units) nor too short. Short primers produce an inaccurate and non-specific DNA amplification product, and long primers result in a slower hybridization rate. On average, the DNA fragment to be amplified should be between 1 and 10 kB in size.
The primer structure must be relatively simple and not contain any internal secondary structure to avoid internal folding. It is also necessary to avoid primer-primer hybridization, which creates primer dimers and disrupts the amplification process. When designing, if you are not sure which nucleotide to place at a certain position within the primer, more than one nucleotide can be included at that position, which is called a mixed site. A nucleotide-based molecular insert (inosine) can also be used instead of a normal nucleotide to achieve broader pairing capabilities.
Taking into account the information above, primers should generally have the following properties:
-
Length of 18-24 bases
-
40-60% G/C content
-
Start and end with 1-2 G/C pairs
-
Melting temperature (Tm) of 50-60°C
-
Primer pairs should have a Tm within a 5 °C difference of each other.
-
Primer pairs should not have complementary regions.
Note: If you are going to include a restriction site at the 5’ end of your primer, keep in mind that a 3 to 6 base pair “clamp” must be added upstream so that the enzyme cleaves efficiently (for example, GCGGCG-restriction site-your sequence).
Primer characteristics
Primer length
Each sense and antisense primer should have a length of between 18-24 bases, since the number of bases influences the specificity of the sequence to be amplified, as long as the annealing temperature is optimal. The length of the primer influences the efficiency of annealing; the longer the primer, the less efficient the alignment. As a smaller amount of primer binds to the template DNA sequence in each cycle, this results in a significant decrease in the amount of amplified product. However, primers should not be too short, since they can bind in a non-specific way. This results in amplification of unwanted products and a decrease in the product of interest.
Specificity
Primers must be specific to delimit the region that is to be amplified. The specificity of the primer depends at least in part on its length. Primers should be chosen so that they have a unique sequence within the DNA to be amplified. Since DNA polymerase can be activated at different temperatures, primer extension occurs at a temperature lower than the annealing temperature. If the temperature is too low, non-specific annealing can occur. The best results are obtained with a melting temperature between 55 and 72ºC, which corresponds to a primer length of between 18 and 24 bases.
Melting temperature (Tm)
The Tm is the temperature at which half of the DNA strands are single-stranded and half are double-stranded. It is desirable for primers to have similar or very close Tm values, with a maximum difference of 5 ºC. If the primers do not have similar Tm values, the amplification is less efficient and may even not be carried out. The primer with the higher Tm works poorly at lower temperatures, and the primer with the lower Tm does not bind at higher temperatures. To avoid these problems during the reaction, the primers are analyzed by means of bioinformatics tools that indicate the Tm of each one of them. Another way to know the Tm is by calculating it using the formula Tm = 4(G+C) + 2 (A+T), this formula gives a good approximation of the Tm value of each primer and is valid for primers of between 18 and 24 bases.
Annealing temperature
Primers must have an annealing temperature of at least 50 ºC. The relationship between annealing temperature and melting temperature is:
The annealing temperature should be 5 ºC lower than the melting temperature (Tannealing = Tm primer – 5 ºC).
This temperature serves as a reference, since it is possible that the annealing temperature determined using this rule is not the appropriate one and several experiments may have to be carried out to determine the optimal temperature of the reaction.
The simplest way to calculate the annealing temperature is with a gradient thermocycler, where several temperatures are tested in a single run and thus the optimal temperature of a particular reaction is determined. A high Tannealing prevents the binding of the primers, and a low Tannealing favors the non-specific binding of the primers. This results in the obtaining of various sizes of amplicons, which at the end of the process are observed in the agarose gel as non-specific bands.
Complementary primer sequences
Regions with the capacity to form internal secondary structures, such as the formation of dimers, complementarity between them or hairpin formation, should be avoided. It is essential that primers do not present sequences with internal homology of more than 3 base pairs. If the primer has zones of self-homology, hairpin structures can form that interfere with annealing to the template DNA. Partial homology in the central regions of the primers can also interfere with alignment. If the homology occurs at the 3’ end of either of the primers, they can form primer dimers, which generally prevent the formation of the desired product through a competition mechanism.
G/C content and stretches of polypyrimidine (T, C) or polypurine (A, G)
The G:C (Guanine:Cytosine) content should be in the range of between 40 and 55%. The greater the number of G and C the primer has, the higher the melting temperature (Tm). On the other hand, poly X sequences (X= G or C or T or A) should also be avoided; the presence of poly G or poly C sequences favors non-specific hybridization. Poly-A and poly-T sequences can reduce the efficiency of amplification, as can stretches of polypyrimidines (T, C) and polypurines (A, G). Ideally, primers should have a random mix of nucleotides, a GC content of 50% and a length of approximately 20 bases. In this way, the Tm will be between 56 – 62 ºC.
Sequence at the 3’ end
An important point is the inclusion of a G or C residue at the 3’ end of the primers. 3’ ends with G/C are suitable because the triple bonds that these bases form favor the efficiency of the reaction. In addition, they minimize the possibility that the double strand formed between the primer and the DNA to be amplified will open.
Primer analysis
A pair of primers that are not properly analyzed can lead to little product, non-specific products or even no product, due to non-specific amplification and/or the formation of primer dimers that compete during the reaction.
To carry out primer analysis, bioinformatics tools are used. With these bioinformatics tools, we can compare the primer sequences against databases and thus characterize them. With the results obtained from the analysis, we determine whether the analyzed primers are suitable or not for carrying out the PCR reaction in the laboratory.
Two of the most used tools, BLAST and Primer-BLAST, these tools are not mutually exclusive, they complement each other for the analysis of the primers.
BLAST
As an example, a pair of primers for the glyceraldehyde-3-phosphate dehydrogenase (gadph) gene of Ovis aries musimon Common mouflon is analyzed.
Below is an example of a sense primer and antisense primer sequence for the gadph gene of Ovis aries musiman:
Sense primer 5'-CCACTGGGGTCTTCACTACC-3'
Antisense primer 5'-AAGCAGGGATGATGTTCTGG-3'
Go to NCBI’s Nucelotide BLAST.
The sequence of the sense primer is transcribed, in the 5’ to 3’ direction, we select the Others option and use the nr/nt nucleotide collection database (Nucleotide collection nr/nt). We click on the BLAST option.
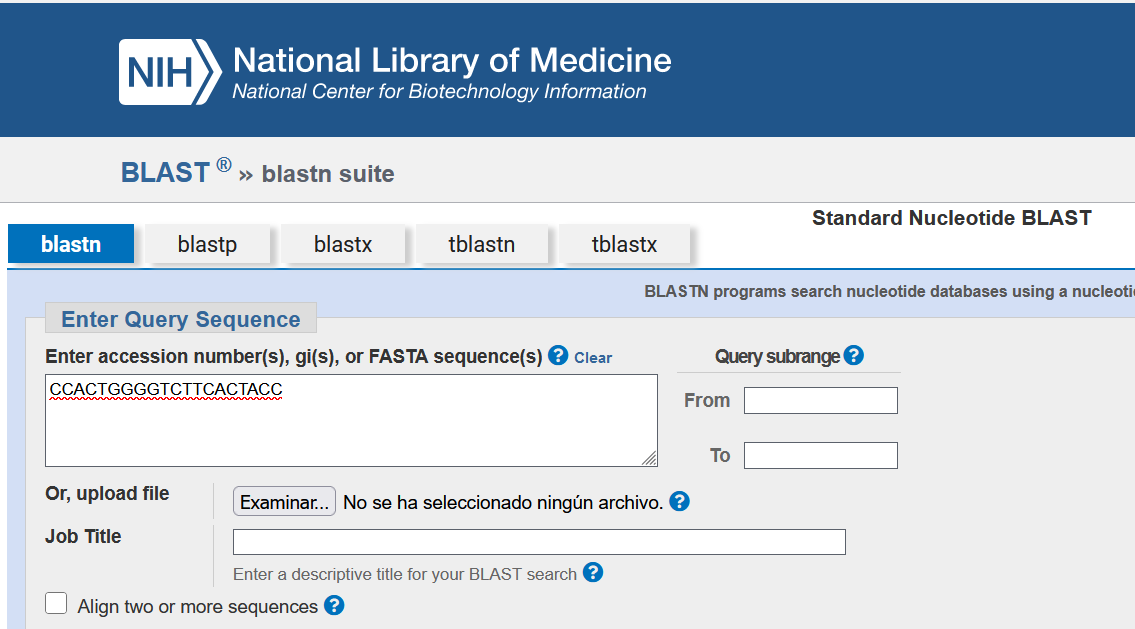
At this point, the sequence is compared against the nr/nt database. The results are displayed with the sequences that BLAST reports as similar (or identical) to the primer being analyzed.
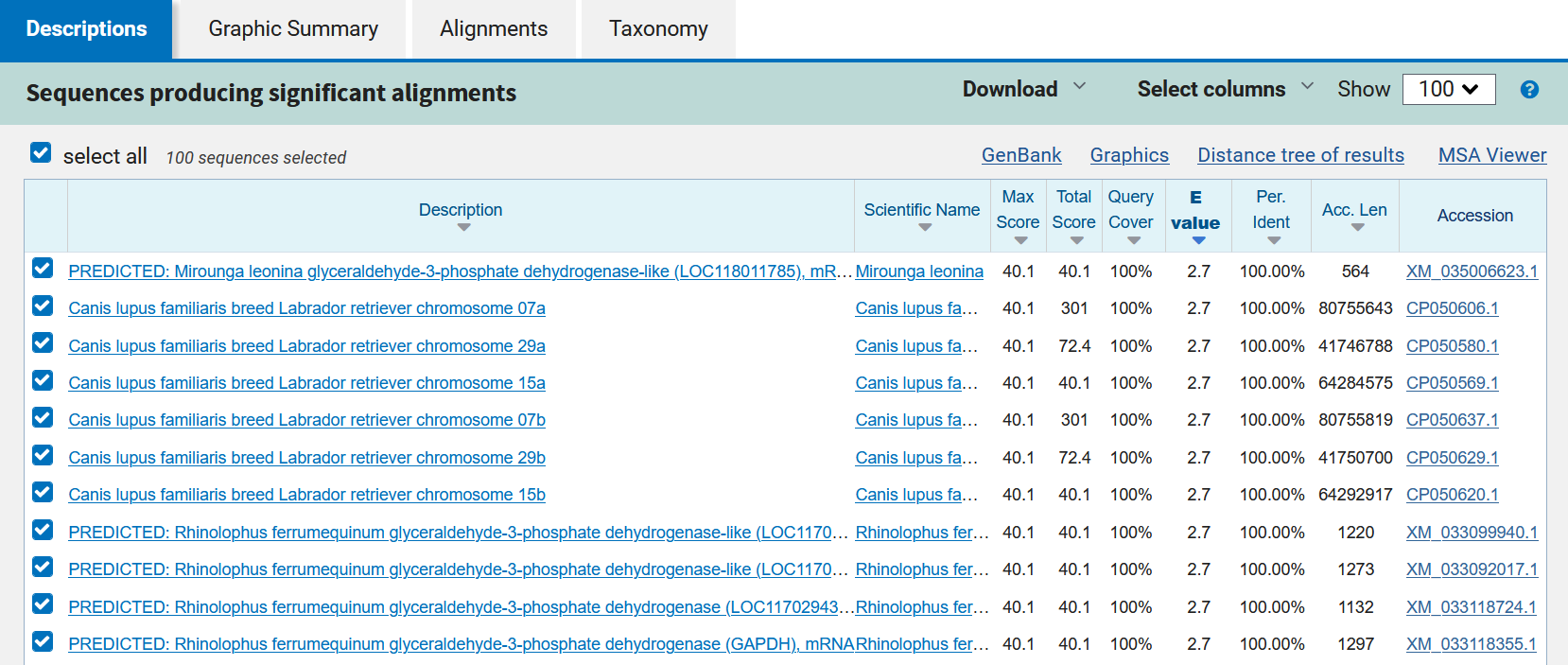
In the example, it is evident that for the sequence that was analyzed, BLAST found several similar sequences. This means that in several species there is a sequence homologous to the sense primer; this sequence may belong to the same protein family that is being analyzed. To see the specificity of the species of interest, the values that the analysis yields are analyzed.
The third section corresponds to the descriptions of the alignments:
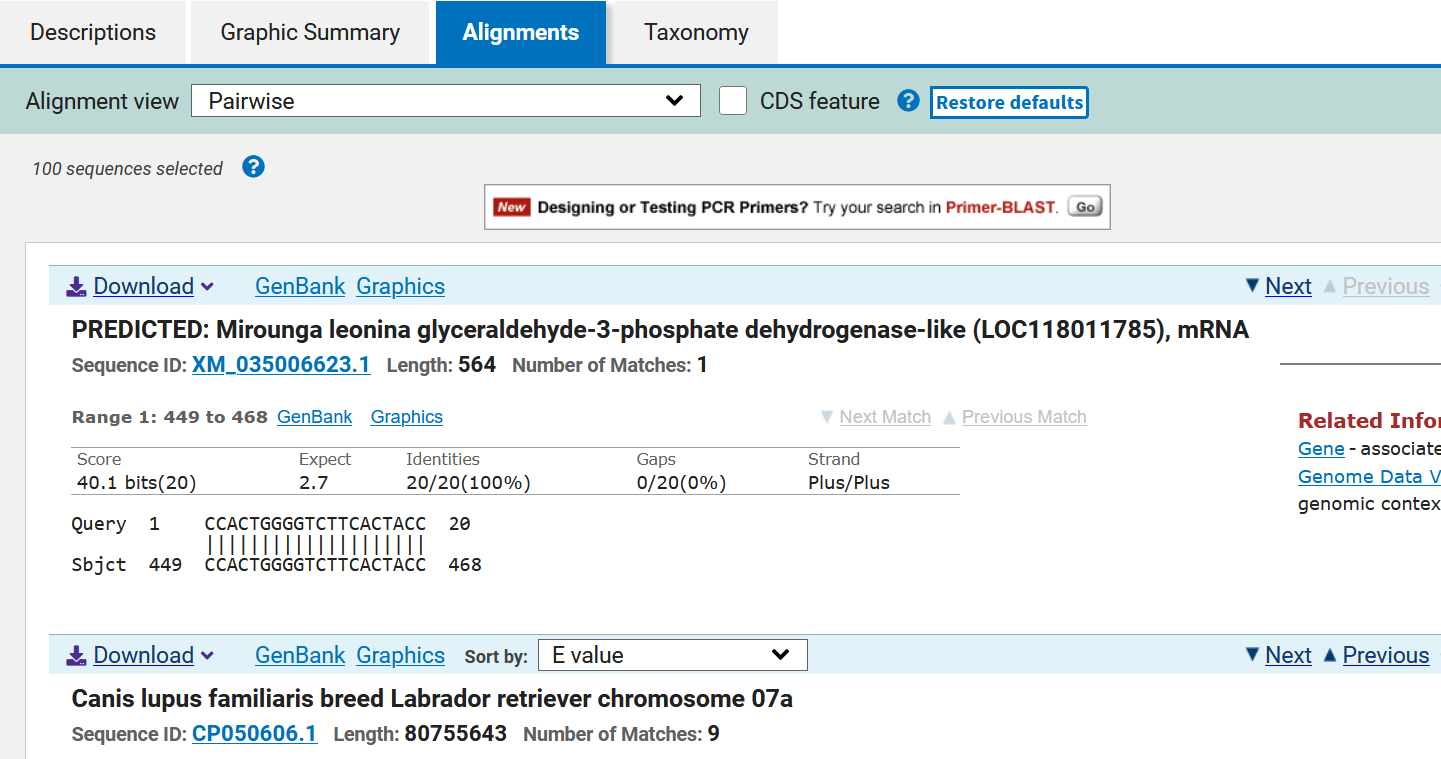
This is a list of the sequences found (ordered according to their E value); each of the alignments is evaluated to determine its statistical significance. The resulting alignments are called High Score Pairs or HSPs, and the cutoff E value or e-value (e-value) allows defining which alignment is suitable according to its statistical significance; the lower the E value, the more significant an alignment is. The E value depends on the database used and the length of the primer sequence. In addition, a series of data is displayed such as the species with which the sense primer sequence matches and the reference of the sequence. The species of interest is searched for among the species and it is verified that it is indeed the gene of interest:
TODO. In our search we did not find ovis aries musiman!
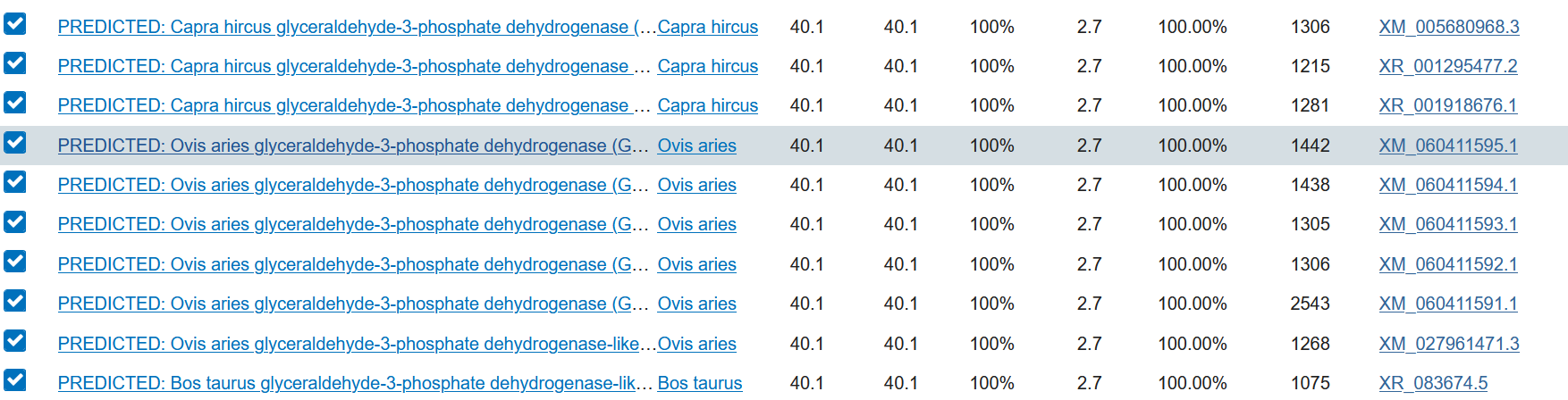
It is important to verify that the reference of the analyzed gene is the same as that reported in the reference article of the sequence. If the sense primer sequence was designed, it will have to be verified that the reference number of the sense primer reported in PubMed matches that of the antisense primer.
For the analysis of the antisense primer, the sequence of that primer is taken, its complementary sequence is elaborated and subsequently the complementary sequence is transcribed from right to left.
TODO Do it with biopython.
Sense primer 5'-CCACTGGGGTCTTCACTACC-3'
Antisense primer 5'-AAGCAGGGATGATGTTCTGG-3'